Historically, IVDs have been treated as low-risk medical devices on the grounds that physicians do not rely on a single piece of information when making diagnoses or treating patients. However, this perception is changing. For example, a recent FDA report on medical device safety states that up to 80% of medical decision-making is now guided by the use of laboratory tests [1]. A leading IVD manufacturer claims that 70% of patient information comes from IVD analysis of blood and other body fluids performed in centralized laboratories [2]. Furthermore, AdvaMed (Washington, DC) and the European Diagnostic Manufacturers Association (EDMA; Brussels) argue that the information from IVD testing has been greatly undervalued, a position supported by recent analyses of the value of laboratory testing [3-5].
While hard data to support such beliefs may be lacking, the FDA Web site has been listing an increasing amount of medical device reporting (MDR) data from IVD manufacturers; the number of Class II IVD recalls, which imply a significant risk to health, has now surpassed Class III IVD recalls; and Class I recalls, which were previously rare in the IVD industry, are occurring more often. Regardless, it would be difficult to argue that IVD assays play an insignificant role in medical decisions and thus present no risk to patients. Furthermore, risk management is not limited to deaths or serious injuries. The burden is on IVD manufacturers to determine and control the potential for any harm through an objective process.
How can IVD manufacturers determine the true risk of their products and comply with regulatory requirements for managing risk? This series of articles will explore the application of medical device risk management principles to IVDs and attempt to place IVD risks in the proper perspective. A systematic risk management process is described in ISO 14971, a voluntary standard developed jointly by the International Organization for Standardization (ISO; Geneva) and the International Electrotechnical Commission (IEC; Geneva) [6]. Part 1 of this series covers regulatory requirements, general risk management principles, and planning and documenting a risk management program.
Исторически было принято рассматривать приборы для лабораторной in vitro диагностики (IVD) как медицинское оборудование группы низкого риска на том основании, что врачи при определении диагноза или лечения пациента не полагаются на информацию только из одного источника. Однако это восприятие меняется. Например, в недавнем докладе FDA о безопасности медицинских устройств говорится, что до 80% принятых медицинских решений в настоящее время основываются на использования именно результатов лабораторных исследований [1]. Ведущий производитель IVD утверждает, что 70% информации о пациенте поступает из IVD анализов крови и других жидкостей организма, которые проводятся в централизованных лабораториях [2]. Кроме того, AdvaMed (Вашингтон, США) и Европейская ассоциации производителей диагностического оборудования (EDMA; Брюссель) утверждают, что информация о значимости результатов, получаемых в клинических лабораториях при IVD исследований, была значительно недооценена [3-5].
Хотя точные данные в поддержку такого утверждения отсутствуют, на сайте FDA в Интернете приведены данные отчетов от производителей IVD медицинских устройств об увеличение числа упоминаний о приборах Класса II риска по сравнению с приборами Класса III риска, как предполагающих существенный риск для здоровья, а число упоминаний о приборах Класса I, которые ранее были редки в отрасли IVD, происходят чаще. Как бы то ни было, трудно утверждать, что IVD анализы играют незначительную роль для медицинских решений и, следовательно, не представляют риска для пациентов. Кроме того, управление рисками не ограничивается выяснением причин смерти или серьезных телесных повреждений. На производителей IVD возлагается бремя посредством объективного процесса определять и контролировать любой потенциальный ущерб посредством объективного процесса.
Как производители IVD приборов могут определить реальный риск их продукции и соблюдать нормативные требования по управлению рисками? В этой серии статей будет рассмотрено применение принципов управления медицинскими рисками по отношению к IVD устройствам и будет сделана попытка поставить IVD риски в надлежащий ряд. Систематический процесс управления рисками описан в стандарте ИСО 14971 – это добровольный стандарт, разработанный совместно с Международной организацией по стандартизации (ИСО, Женева) и Международной электротехнической комиссии (МЭК, Женева) [6]. Часть 1 этой серии статей охватывает нормативные требования, общие принципы менеджмента риска, а также планирование и документирование программы менеджмента риска.
Regulatory Aspects
Нормативные аспекты
ISO 14971 has been adopted by most countries as the preferred standard for complying with risk management requirements for medical devices. FDA recognizes the standard and participates in the technical committee that developed ISO 14971. In the European Union, a declaration of conformity to ISO 14971 creates a presumption of compliance with the IVD Directive’s essential requirements pertaining to risk management. Australia, Canada, Japan, and several other countries have adopted the ISO 13485 quality system, which requires risk management. Given ISO 14971’s flexibility, logical framework, and strong support from the regulatory community, adopting a different risk management approach would be a questionable strategy.
While risk analysis is the term that FDA uses to describe one of the design control requirements in the quality system regulation (QSR), the theme in regulatory and quality circles today is risk management and its integration into the quality management system.7 According to ISO 14971, risk analysis is only the first step in a total product life cycle risk management process. Risk management is a much broader concept, and a recent guidance from the Global Harmonization Task Force (GHTF) stresses its integral role in the quality management system [8].
The difference between the risk analysis required in 1996 and today’s risk management expectations is more an evolution of terminology than an escalation of requirements. When FDA drafted the QSR, risk analysis and risk assessment were the terms used, often interchangeably, to describe the activities that IVD manufacturers undertake to minimize the risk of using their products. In the meantime, ISO defined risk concepts more precisely for medical devices. The QSR’s true regulatory intent is revealed in its preamble, which indicates that FDA never intended manufacturers to stop the risk management process after only the first stage.
While the risk management requirements in the European IVD Directive are often thought to be more demanding, in reality they are not very different from the expectations laid out in the QSR preamble.
When their risk management program is audited, many IVD manufacturers still point to a risk analysis document prepared during product development as their only evidence of risk management. Nearly 10 years have passed since FDA first published the QSR. The third edition of ISO 14971 with an expanded set of IVD guidelines is nearing completion. And the GHTF has published a final guidance on integrating risk management into the quality system [8, 9]. A review of the basic requirements of a risk management program for IVDs is therefore timely.
ИСО 14971 был принят в большинстве стран в качестве предпочтительного стандарта для выполнения требований менеджмента риска медицинского оборудования. FDA признает стандарт и участвует в работе технического комитета по развитию ISO 14971. В странах Европейского Сообщества декларирование соответствия ISO 14971 подтверждает соответствие основным требованиям директивы IVD, связанным с управлением рисками. Австралия, Канада, Япония и ряд других стран приняли стандарт ISO 13485, содержащий требования к системам менеджмента качества предприятий, производящих медицинские изделия, этот стандарт требует наличия менеджмента риска. Учитывая гибкости ISO 14971, его логические рамки, а также сильную поддержку со стороны регулирующих органов, принятие другого подхода к управлению рисками было бы сомнительным.
Хотя анализ рисков является термином, который FDA использует для описания одного из требований для контроля параметров конструкции в Правилах системы качества (QSR), в нормативных кругах этот термин сегодня означает менеджмент риска и его интеграцию в систему менеджмента качества [7]. В соответствии с ISO 14971, анализ рисков является только первым шагом в рамках полного жизненного цикла менеджмента риска изделия. Менеджмент риск является гораздо более широким понятием, и недавно изданное руководство целевой группы по глобальной гармонизации (GHTF) подчеркивает его роль в системе менеджмента качества [8].
Разница между анализом риска в соответствии с требованиями 1996 год и ожидаемыми сегодня результатами менеджмента риска больше проявляется в изменении терминологии, чем в ужесточении требований. Когда FDA разработало QSR, анализа риска и оценки риска были терминами часто взаимозаменяемы для описания деятельность, которую производители IVD изделий обязуются свести к минимуму риск при использовании их продукции. В то же время, для медицинского оборудования ISO определяет концепцию риска более точно. Истинные нормативы правил QSR раскрываются в их преамбуле, в которой указывается, что FDA никогда не указывало производителям ограничить процесс менеджмента риска всего лишь первым этапом.
Хотя требованиям менеджмента риска Европейской директивы IVD часто считаются более жесткими, в действительности они не очень отличаются от указаний, изложенных в преамбуле к QSR.
При проведении аудита программы менеджмента риска многие производители IVD изделий все еще указывают на анализ рисков, подготовленный в ходе разработки продукта, как на единственное доказательство по управлению рисками. Почти 10 лет прошло с тех пор, как FDA впервые опубликовало QSR. Третье издание ИСО 14971 с расширенным перечнем руководства относительно IVD близится к завершению. И GHTF опубликовала окончательные рекомендации по интеграции менеджмента риска в систему качества [8, 9]. Поэтому обзор основных требований программе менеджмента риска для IVD изделий является своевременным.
Risk Management Process
Процесс управления рисками
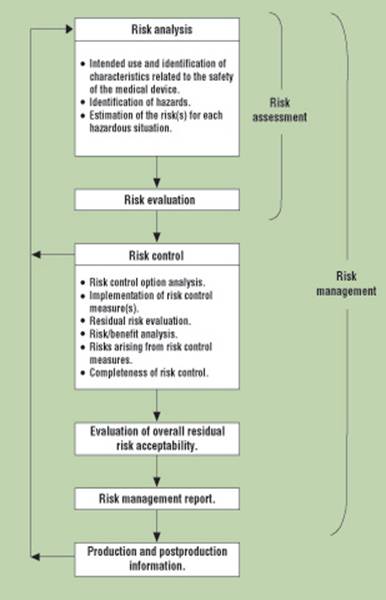
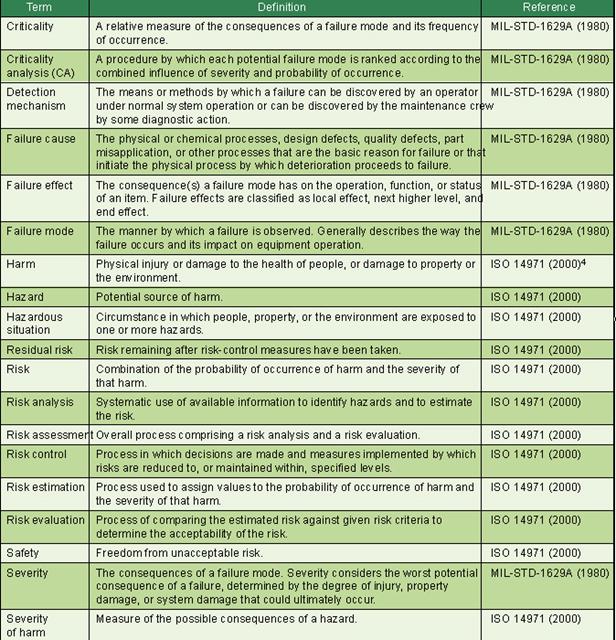
Fig. 1. A schematic representation of the risk management process. (Source: ISO/DIS 14971:2005).
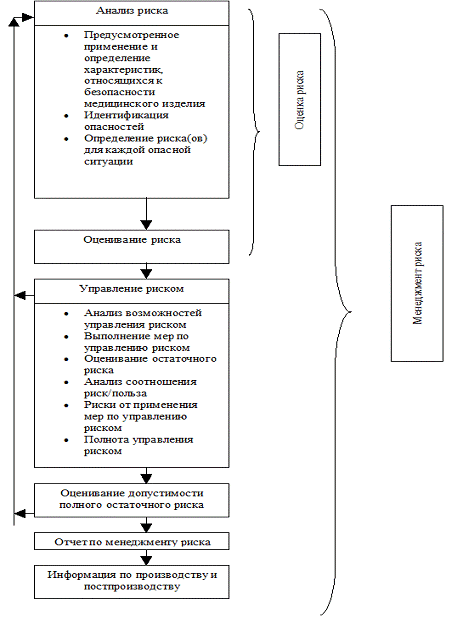
Рис. 1. Схематическое изображение процесса управления рисками. (Источник: ISO / DIS 14971:2005).
According to ISO 14971, a systematic risk management process for medical devices, including IVDs, involves the following four main stages (see Figure 1):
• Identifying hazards inherent in the use of IVD products, and estimating the risks of harm to patients, laboratory workers, and the environment.
• Evaluating the acceptability of such risks against criteria established by company management.
• Reducing risks to a level that complies with the company’s risk acceptability policy, and verifying that the risk controls are effective.
• Monitoring internal and external product experience for new hazards, increased risks, and the possibility that society’s tolerance for risks has changed.
The first three stages integrate logically into the flow of a product design and development process [10]. The last stage of risk management is ongoing throughout the life of a product, and overlays the entire quality system architecture. The vigilance required to sustain risk management involves almost every organization in an IVD company, and it is a never-ending responsibility of top management.
The GHTF member countries view risk management as a necessary part of an effective quality management system. In its recent guidance document, GHTF takes the position that integrating risk management efforts into the quality management system is necessary to ensure that such efforts will be coordinated and all identified risk issues will be brought to closure.
С целью соблюдения нормативных требований, актуальными считаются только риски, связанные с воздействием на здоровье, безопасности или экологической опасности. Важные для компании-производителей IVD изделий бизнес-риски, связанные с задержками в осуществлении проектов, неэффективностью производственных процессов, или недовольством клиенты, не имеют большого интереса для FDA. Если завод-изготовитель все же рассматривает коммерческие риски в общей оценке рисков, документация должна четко указать, что эти риски считаются вторичными по сравнению с любыми рисками, связанными со здоровьем или безопасностью. В идеале, производителям IVD изделий следует отдельно рассмотреть бизнес-риски.
Согласно ISO 14971, систематический процесс менеджмента риска для медицинских приборов, в том числе IVD изделий, включает в себя следующие четыре основных этапа (см. Рисунок 1):
• Определение опасностей, связанных с использованием изделий IVD, и оценки рисков причинения вреда пациентам, работникам лабораторный и окружающей среде.
• Оценка приемлемости такого риска в отношении критериев, установленных руководством компании.
• Снижение рисков до уровня, который соответствует приемлемому риску с точки зрения политики компании, и проверка того, что управление риском являются эффективным.
• Мониторинг внутреннего и внешнего опыта использования изделий в отношении появления новых опасностей, возрастания рисков и изменения требования общества к рискам.
Первые три этапа логично интегрируются с процессами разработки и конструирования изделий [10]. Последний этап управления рисками действует на протяжении всей жизни продукта и вписывается в архитектуру системы качества. Почти каждая организация производитель IVD изделий осознает необходимость процесса поддержания управление рисками, и с точки зрения ответственности топ-менеджмента этот процесс является бесконечным.
В странах членах GHTF управление риском рассматривается как необходимый элемент эффективной системы управления качеством. Позиция, занятая руководством GHTF В своем недавнем документе, заключается в том, что интеграция усилий по управлению рисками в системе управления качеством является необходимым условием обеспечения того, чтобы такие усилия будут координироваться и все вопросы по выявленным рискам будут рассмотрены и закрыты.
Связь между понятиями риска
IVD manufacturers need to understand the potential of their assays to cause or contribute to patient harm. This determines the level of risk, which depends on the severity of the possible harm and the probability that actual harm will occur. An important concept clarified in the new revision of ISO 14971 is that harm can only occur when a patient is placed in a hazardous situation, which means exposure to the hazard. For IVDs, this generally means that an incorrect result (the hazard) must be reported to a physician or other healthcare provider in a position to act on the result (the hazardous situation). It may also mean that a critical result was not able to be delivered when needed.
An IVD assay has inherent risk if physicians rely on the results for medical decisions. Obviously, IVD assays that fail to meet performance specifications can be hazardous. However, a state-of-the-art assay that conforms to all performance specifications can also generate hazardous results. Given the biological nature of many IVD assay components, state-of-the-art specificity is rarely capable of perfect discrimination between positive and negative specimens, and some well-established quantitative methods operate at the limit of their accuracy. For example, the National Institute of Standards and Technology (NIST; Gaithersburg, MD) commissioned a report that quantified the effects of misdiagnosis from incorrect calcium results from state-of-the-art clinical test methods. Such misdiagnoses resulted in $119 million in extra costs to the U.S. healthcare system, in part due to unnecessary medical procedures performed in response to falsely abnormal (i.e., potentially hazardous) results[11].
On the other hand, an incorrect result may be a hazard, but it may not necessarily present a significant risk. The likelihood that a patient will be exposed to harm may be reduced by the ability of the IVD instrument itself, or laboratory workers or even physicians, to detect hazardous test results. According to ISO 14971, IVD manufacturers must formulate a preestablished risk acceptability policy that enables management to decide whether risks from incorrect or delayed results are acceptable. Factors such as patient benefits, applicable regulations and standards, technology state of the art, and current values of society dictate the acceptability of the risks.
Неудачи в идентификации и управлении рисками зачастую можно объяснить отсутствием понимания основных концепций риска, и недопонимание между ведомствами. Как стандарт ISO, так и руководство GHTF строго подчеркивают, что производители IVD устройств должны использовать единую терминологию риска для ясного понимания взаимосвязи различных видов риска в рамках своих организаций. Использования одного языка также может помочь при объяснении внешним аудиторам или контролерам FDA как компания проводит менеджмент риска.
Производители IDV устройств должны понимать, какой потенциальный вред результаты лабораторных анализов могут оказать пациенту. Это определяет степень риска, который зависит от тяжести возможного ущерба и вероятности того, что фактический ущерб произойдет. Важно уточнить, что концепцией новой редакции стандарта ИСО 14971 является то, что вред может произойти, когда пациент находится в опасной ситуации, которая приведет к опасным воздействиям. Для IVD изделий это обычно означает, что неправильный результат (опасность) сообщен врачу или другому медработнику, которые примут решение на основании этого результата результат (опасной ситуации). Это также может означать, что важные результаты лабораторных исследований мог оказаться полученными с запозданием.
Результатам IVD исследований всегда присущи риски, если врачи полагаются на результаты для медицинских решений. Очевидно, что результаты IVD исследований, не отвечающие техническим требованиям могут быть опасными. Однако даже самые современные методы клинических исследований, которые соответствует всем техническим требованиям, также могут привести к опасным результатам. Учитывая биологическую природу многих компонентов IVD исследований, даже современные методы клинических исследований редко способны точно разграничить положительные и отрицательные пробы, а некоторые даже хорошо известные количественные методы работают на пределе их точности. Например, Национальный институт стандартов и технологии США (NIST; Gaithersburg, MD) подготовила доклад о количественных последствиях ошибочного диагноза из-за неправильных результатов определения кальция современными клиническими методами. Такие ошибочные диагнозы привели к дополнительным расходам в 119 миллионов долларов в системе здравоохранения США. Отчасти это произошло из-за выполнения ненужных медицинских процедур в ответ на ложные, ненормальные (то есть потенциально опасные) результаты [11].
С другой стороны, неправильный результат может быть опасным, но это не обязательно может представлять значительный риск. Вероятность того, что пациент будет подвергаться вредному воздействию, может быть уменьшена способностью самого IVD устройства, или лаборанта или даже врачи обнаружить опасные результаты клинических исследований. Согласно ISO 14971, производители IVD изделий должны разрабатывать политику предустановленной приемлемости риска, что позволяет решать, какие риски, связанные с неправильными результатами или задержкой результатов являются приемлемыми. Такие факторы, как польза для пациента, применимые правила и стандарты, современные технологии, и современные общественные ценностей диктуют требования по приемлемости риска.